Age-related hearing loss is common in those persons over 65 years of age. Nearly 33% of persons over 65 years of age experience this type of hearing loss. Age-related hearing loss is typically a high-frequency loss that occurs progressively but slowly throughout adulthood. While age-related hearing loss begins in the high frequencies it slowly progresses to lower frequencies with time. This study examined the relationship between age-related hearing loss and anxiety in those over 65 years of age.
Sixty-seven persons with age-related hearing loss and 68 normal-hearing controls participated in this cross-sectional study. The criteria to be included in the age-related hearing loss group was a four-frequency pure tone average of >25 decibels hearing level in the better hearing ear. All participants had three-dimensional magnetic resonance imaging (MRI), pure tone audiometric testing, and anxiety and depression scales.
The findings included a decrease in grey matter volume in 20 brain regions in the age-related hearing loss group. In addition, a positive correlation was found between high-frequency pure tone average and anxiety scores in the age-related hearing loss group. No relationships were found between depression and either gray matter volume or high-frequency hearing loss.
Ma, W., Zhang, Y., Li, X., Liu, S., Gao, Y., Yang, J., Xu, L., Liang, H., Ren, F., Gao, F., & Wang, Y. (2022). High-Frequency Hearing Loss Is Associated with Anxiety and Brain Structural Plasticity in Older Adults. Frontiers in aging neuroscience, 14, 821537. https://doi.org/10.3389/fnagi.2022.821537
Turner syndrome is a genetic disorder that affects individuals assigned female at birth, characterized by the complete or partial absence of one X chromosome. The condition occurs in approximately 1 in 2,000 to 2,500 live female births and is associated with a distinct set of physical and developmental features.
The pathophysiology of Turner syndrome stems from the haploinsufficiency of genes normally present on both X chromosomes. This genetic imbalance leads to various developmental abnormalities, particularly affecting growth, ovarian function, and cardiovascular development. The loss of specific genes, such as the SHOX gene, contributes to short stature, while the absence of genes involved in ovarian development results in gonadal dysgenesis and infertility in most cases.
Genetically, Turner syndrome can manifest in several ways. The most common karyotype is 45,X, where one X chromosome is completely missing. However, mosaic forms (45,X/46,XX) and structural abnormalities of the X chromosome, such as isochromosome Xq or ring X chromosomes, are also observed. These genetic variations can influence the severity and presentation of the syndrome.
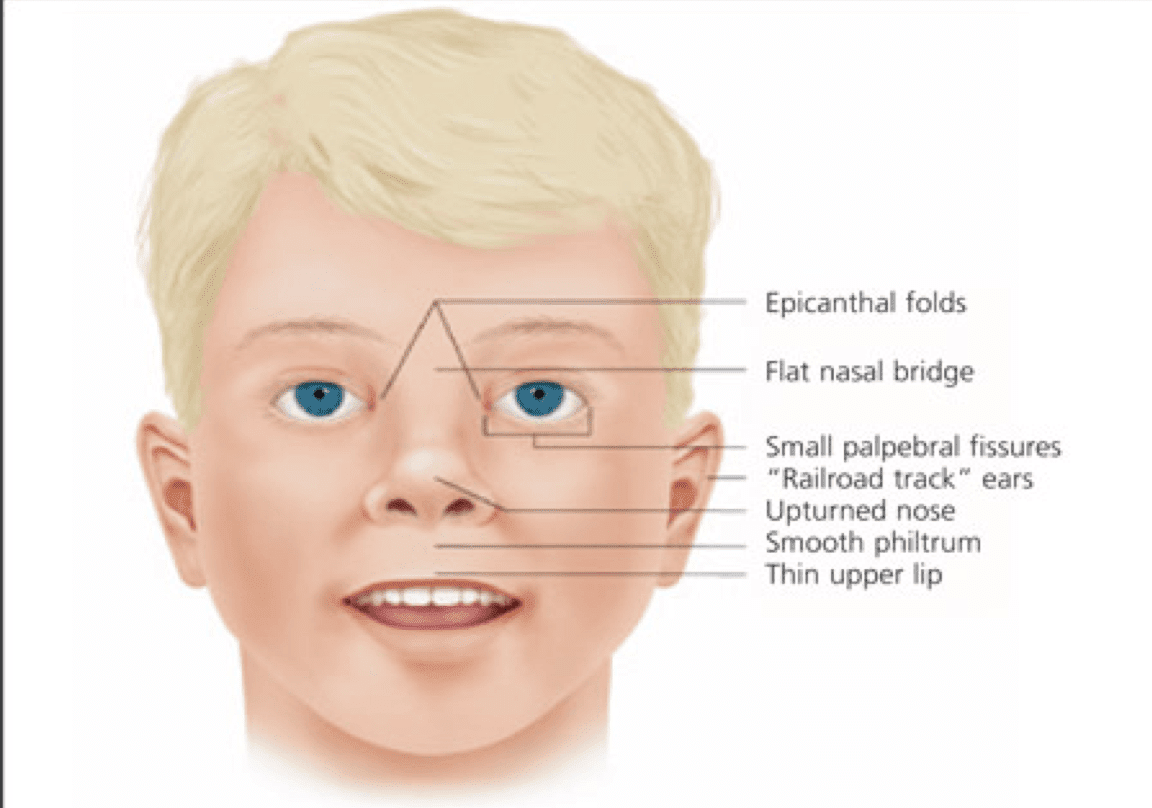
The signs and symptoms of Turner syndrome are diverse and can vary significantly among affected individuals. Common features include short stature, which becomes evident by age 5, and ovarian insufficiency, leading to delayed or absent puberty. Other characteristics may include a webbed neck, low hairline, broad chest with widely spaced nipples, and lymphedema of the hands and feet. Cardiovascular abnormalities, particularly coarctation of the aorta and bicuspid aortic valve, are present in up to 50% of cases. Renal anomalies, hearing loss, and autoimmune disorders are also more prevalent in this population. Cognitive function is generally normal, although specific learning difficulties, particularly in visuospatial and mathematical domains, are common.
People with Turner’s syndrome can have many complications related to genetic abnormalities and can impact nearly every body system.
Heart problems: Many infants with Turner syndrome are born with heart defects or abnormalities in heart structure. Congenital heart defects: Approximately 50% of people with Turner syndrome are born with structural heart problems. The most common heart defect in Turner syndrome is the bicuspid aortic valve, where the valve controlling blood flow from the heart to the aorta has only two flaps instead of the usual three. In the coarctation (narrowing) of the aorta, the aorta is too narrow, causing the heart to pump harder. The aorta can become wider than usual, increasing the risk of rupture (aortic dissection). When the aortic valve is insufficient or too narrow, it restricts blood flow and increases pressure on the left side of the heart. Patients with Turner syndrome may also have high blood pressure can increase the risk of complications from other heart defects.
Hearing loss: This is common and can be due to gradual nerve function loss or frequent middle ear infections. At least 25% of adults with Turner syndrome require hearing aids. Hearing loss tends to progress more rapidly than in the general population and can be either conductive hearing loss or sensorineural hearing loss. Sensorineural hearing loss affects over 50% of adults with Turner syndrome. The combination of mid-frequency dip and early high-frequency loss often necessitates hearing aid use earlier than in the general population
Vision problems: There is an increased risk of strabismus (weak eye muscle control), nearsightedness, and other vision issues. Those with Turner syndrome have high rates of ametropia (refractive errors) are common, affecting around 40% of individuals with Turner syndrome. This includes myopia, hyperopia, and astigmatism. Strabismus is very common, affecting 13-33% of those with Turner syndrome. Esotropia appears to be more common than exotropia. Amblyopia occurs in 16-30% of individuals with Turner syndrome. This is likely related to the high rates of strabismus and refractive errors. About 8% have color vision abnormalities, which is similar to rates in males rather than females in the general population. there are other vision issues that could impact people with Turner syndrome such as keratoconus, cataracts, glaucoma, or uveitis. Vision issues often begin in childhood but may not be recognized early without screening. The high rates of vision problems, especially strabismus and amblyopia, highlight the importance of early and regular ophthalmological screening for individuals with Turner syndrome. Early detection and intervention is crucial to prevent long-term visual impairment. The vision issues do not appear to be strongly associated with any particular karyotype of Turner syndrome.
Kidney problems: Malformations of the kidneys are associated with Turner syndrome, which may increase the risk of urinary tract infections. Approximately 30-33% of individuals with Turner syndrome have kidney abnormalities. Several types of kidney issues could impact persons with Turner syndrome, including horseshoe kidney the most common renal anomaly, occurring in about 71% of those with kidney issues. Malrotation of the kidneys, single kidney, duplex collecting system,
Progression and complications:
Many kidney abnormalities are asymptomatic and do not cause serious medical problems.
However, these abnormalities can increase the risk of:
Urinary tract infections (UTIs)
Hypertension (high blood pressure)
Renal parenchymal damage
Chronic kidney disease (rare)
Detection and diagnosis:
Often discovered incidentally during routine screening
Some cases are diagnosed following recurrent UTIs
A small percentage may be detected prenatally
Autoimmune disorders: There is an increased risk of hypothyroidism, diabetes, celiac disease, and inflammatory bowel disease.
Skeletal problems: These include an increased risk of scoliosis, kyphosis, and osteoporosis.
Learning disabilities: While intelligence is usually normal, there is an increased risk of learning disabilities, particularly with spatial concepts, math, memory and attention.
Mental health issues: Girls and women with Turner syndrome may face challenges in social situations and have an increased risk of anxiety, depression, and ADHD.
Infertility: The majority of women with Turner syndrome experience premature ovarian failure. The ovaries develop normally at first, but egg cells usually die prematurely, and most ovarian tissue breaks down before birth or during childhood. Many girls with Turner syndrome do not undergo puberty naturally and require hormone replacement therapy to initiate pubertal development. Most women with Turner syndrome (>90%) are unable to become pregnant naturally due to the lack of functioning ovaries and egg cells.
Pregnancy complications: For the small percentage who can become pregnant, there are increased risks during pregnancy. A small percentage (2-8%) of women with Turner syndrome, particularly those with mosaic karyotypes, may retain some ovarian function and experience spontaneous pregnancies. However, these pregnancies have higher risks of miscarriage and chromosomal abnormalities in the fetus.
These complications highlight the importance of comprehensive, multidisciplinary care for individuals with Turner syndrome throughout their lives.
Treatment for Turner syndrome is multifaceted and aims to address the various aspects of the condition. Growth hormone therapy is a cornerstone of management, typically initiated in early childhood to improve final adult height. Estrogen replacement therapy is crucial for inducing puberty and maintaining secondary sexual characteristics, bone health, and cardiovascular health. Regular screening for associated conditions, including cardiovascular, renal, and thyroid abnormalities, is essential. Psychological support and educational interventions may be necessary to address learning difficulties and psychosocial challenges.
Recent research has focused on optimizing treatment strategies and understanding the long-term outcomes of Turner syndrome. A study by Fiot et al. (2021) highlighted the importance of early growth hormone treatment in improving adult height outcomes. Cardiovascular management has been emphasized in recent guidelines, with Mortensen et al. (2023) stressing the need for lifelong cardiac surveillance. Advances in reproductive technologies have also opened new possibilities for fertility preservation and treatment, as discussed by Grynberg et al. (2020). The psychological aspects of Turner syndrome, including quality of life and neurocognitive outcomes, have been explored by Cardoso et al. (2022), emphasizing the need for comprehensive care beyond medical management. Lastly, Gravholt et al. (2021) provided updated clinical practice guidelines for the care of girls and women with Turner syndrome, encompassing a lifespan approach to management.
References
Fiot, E., et al. (2021). “Long-term effect of early growth hormone treatment on adult height in Turner syndrome.” European Journal of Endocrinology, 184(1), 1-10.
Mortensen, K. H., et al. (2023). “Cardiovascular health in Turner syndrome: Current knowledge and future directions.” Journal of Clinical Endocrinology & Metabolism, 108(3), 529-543.
Grynberg, M., et al. (2020). “Fertility preservation in Turner syndrome: A comprehensive review and practical guidelines.” Journal of Clinical Medicine, 9(8), 2468.
Cardoso, G., et al. (2022). “Neurocognitive and psychosocial outcomes in Turner syndrome: A systematic review.” Neuroscience & Biobehavioral Reviews, 132, 324-336.
Gravholt, C. H., et al. (2021). “Clinical practice guidelines for the care of girls and women with Turner syndrome: Proceedings from the 2021 International Turner Syndrome Meeting.” European Journal of Endocrinology, 184(3), G1-G68.
Choosing the right hearing aids is an important decision that should be made in consultation with an audiologist or hearing healthcare professional. Here are some steps to consider when choosing hearing aids:
Get a Hearing Evaluation: The first step is to have a comprehensive hearing evaluation conducted by a qualified audiologist. This will help determine the extent and type of hearing loss you have.
Discuss Your Lifestyle and Needs: Talk to your audiologist about your lifestyle and specific hearing needs. Consider factors like your daily activities, social interactions, and the environments where you spend the most time.
Choose the Style: Hearing aids come in various styles, including behind-the-ear (BTE), in-the-ear (ITE), in-the-canal (ITC), and completely-in-the-canal (CIC). The choice of style often depends on your preferences and the degree of hearing loss. You can also use this Hearing Aid Wizard from hearingtracker.com
Technology Level: Hearing aids come in different technology levels, ranging from basic to advanced. High-level technology often offers features like noise reduction, directional microphones, and Bluetooth connectivity. Your choice will depend on your budget and specific needs.
Consider Special Features: Think about any special features that may benefit you. For example, some hearing aids have telecoil options for better phone compatibility, while others are rechargeable for convenience.
Trial Period: Many hearing aid providers offer trial periods that allow you to try the devices and see how they work in different situations. This is essential to determine if the hearing aids meet your expectations.
Ask About Warranty and Support: Inquire about the warranty and ongoing support provided by the manufacturer or hearing aid provider. This can include repairs, adjustments, and follow-up appointments.
Cost and Insurance: Discuss the cost of the hearing aids and inquire about any insurance coverage or financial assistance programs that may be available to you.
Maintenance and Cleaning: Understand the maintenance and cleaning requirements for the hearing aids. Proper care can prolong their lifespan and performance.
Realistic Expectations: Keep in mind that hearing aids can improve your hearing, but they may not fully restore it. It’s important to have realistic expectations about the benefits they can provide.
Ask for Recommendations: If you know someone who wears hearing aids, ask for their recommendations and experiences. Word-of-mouth referrals can be valuable.
Stay Informed: Stay informed about the latest advancements in hearing aid technology and research. Technology is continually evolving, and new options may become available that better suit your needs.
Follow-up: After getting hearing aids, attend follow-up appointments with your audiologist for adjustments and to address any concerns or issues you may have.
Remember that selecting the right hearing aids is a personalized process. What works for one person may not work for another. Your audiologist is your best resource for guidance and support throughout this journey, so be sure to ask questions and share your concerns openly.
As a farmer, you’re exposed to a variety of loud noises on a daily basis – from tractors and combines to livestock and grain dryers. While these sounds may seem like an unavoidable part of farm life, they pose a serious risk to your hearing health. This article aims to highlight the dangers of noise-induced hearing loss (NIHL) in agriculture, its long-term consequences, and practical ways to protect your hearing.
The Dangers of Noise-Induced Hearing Loss in Agriculture
Farming is consistently ranked as one of the occupations with the highest risk for hearing loss. A study by Lie et al. (2016) found that farmers have a 38% higher risk of hearing loss compared to other occupations. The primary culprit is prolonged exposure to high noise levels from farm machinery and equipment.
Many common farm activities produce noise levels well above the safe threshold of 85 decibels (dB).
Most people’s ears ring after they are exposed to loud noises. The ringing or tinnitus can sound a bit different to different people, a ring like a bell, a buzzing like bees, or just noise that doesn’t go away. Audiologists measure how loud a sound is in units called decibels (dB). Hearing loss can happen due to repeated exposure over time (years usually). However, hearing loss can occur due to a one-time very loud noise exposure of 120 dB. Think of 120 dB as how loud an indoor basketball stadium is when everyone is cheering, rock concerts, or jet planes at the ramp. The table below was taken from Penn State Extension, and describes what sounds are too loud and how long you can go daily without hearing protection without damage. Remember though, this is what we know now, and can change as we learn more about how our ears work and heal.
Exposure to these noise levels for extended periods can cause permanent damage to the delicate structures in your inner ear, leading to noise-induced hearing loss.
Long-Term Consequences of Noise-Induced Hearing Loss
The effects of NIHL extend far beyond just difficulty hearing. Recent research has highlighted several long-term consequences:
A study by Curhan et al. (2019) found that hearing loss is associated with accelerated cognitive decline and an increased risk of dementia. Mid-life hearing loss doubles the risk of developing dementia compared to any other single risk factor. Poor hearing performance is significantly associated with lower cognitive function in cross-sectional and longitudinal analyses. Hearing impairment is associated with brain atrophy in regions important for auditory processing and cognition, including the temporal gyrus, hippocampus, amygdala, precuneus, and prefrontal cortex. People with age-related hearing impairment show increased levels of phosphorylated tau in cerebrospinal fluid, a biomarker associated with Alzheimer’s disease. Hearing loss may lead to social withdrawal, reducing cognitive stimulation. The brain may reallocate cognitive resources to process degraded auditory input, leaving fewer resources for other cognitive tasks. There may be underlying factors contributing to hearing loss and cognitive decline.
Difficulty communicating can lead to social withdrawal and isolation, potentially impacting mental health. Impaired hearing can make it challenging to detect warning signals or approaching vehicles, increasing the risk of farm-related accidents.
Impact of hearing aids on cognitive function
Some studies suggest that using hearing aids may help maintain better cognitive function over time compared to those who don’t use them. A recent clinical trial found that hearing aids reduced the rate of cognitive decline by almost 50% over three years in older adults at high risk of dementia. Hearing aids and other interventions can be costly, and severe hearing loss may impact your ability to work efficiently. Hearing loss can significantly reduce overall quality of life, affecting relationships and daily activities.
Protecting Your Hearing on the Farm
The good news is that NIHL is largely preventable. Here are some effective strategies to protect your hearing:
Use Hearing Protection: Always wear earplugs or earmuffs when operating loud machinery. A study by McCullagh et al. (2016) found that consistent use of hearing protection devices can significantly reduce the risk of hearing loss among farmers.
Limit Exposure Time: Follow the 60/60 rule – if you must be exposed to loud noise, limit it to 60 minutes and then take a 60-minute break in a quieter environment.
Maintain and Upgrade Equipment: Regular maintenance can help reduce noise levels. When possible, invest in quieter, modern equipment.
Create Quiet Zones: Designate areas on your farm for noise-free breaks.
Get Regular Hearing Check-ups: Annual hearing tests can help detect NIHL early when interventions are most effective.
Education and Training: Participate in hearing conservation programs. Cramer et al. (2018) found that targeted education can improve farmers’ use of hearing protection.
Types of hearing protection that can be used when a person is in loud environments.
Conclusion
Your hearing is a precious asset that, once damaged, cannot be fully restored. By understanding the risks of NIHL and implementing protective measures, you can preserve your hearing health and ensure a long, productive farming career. Remember, protecting your hearing is not just about maintaining your ability to hear – it’s about safeguarding your overall health, safety, and quality of life.
References
Lie, A., Skogstad, M., Johannessen, H. A., Tynes, T., Mehlum, I. S., Nordby, K. C., … & Tambs, K. (2016). Occupational noise exposure and hearing: a systematic review. International archives of occupational and environmental health, 89(3), 351-372.
Curhan, S. G., Willett, W. C., Grodstein, F., & Curhan, G. C. (2019). Longitudinal study of hearing loss and subjective cognitive function decline in men. Alzheimer’s & Dementia, 15(4), 525-533.
McCullagh, M. C., Banerjee, T., Yang, J. J., Bernick, J., Duffy, S., & Redman, R. (2016). Gender differences in use of hearing protection devices among farm operators. Noise & health, 18(82), 168.
Cramer, M. E., Wendl, M. J., Sayles, H., Duysen, E., & Achutan, C. (2018). Knowledge, attitudes, and practices for respiratory and hearing health among Midwestern farmers. Public Health Nursing, 35(4), 308-315.
Feder, K., Michaud, D., McNamee, J., Fitzpatrick, E., Davies, H., & Leroux, T. (2017). Prevalence of hazardous occupational noise exposure, hearing loss, and hearing protection usage among a representative sample of working Canadians. Journal of occupational and environmental medicine, 59(1), 92-113.
Neitzel, R. L., Andersson, M., & Andersson, E. (2016). Comparison of multiple measures of noise exposure in paper mills. The Annals of occupational hygiene, 60(5), 581-596.
Usher syndrome is an inherited condition that causes both hearing loss and progressive vision loss due to retinitis pigmentosa (RP). It is the most common cause of combined deafness and blindness.
Pathophysiology Usher syndrome results from mutations in genes involved in the development and function of specialized cells in the retina and inner ear. These mutations lead to abnormal development or degeneration of hair cells in the inner ear, causing hearing loss and progressive degeneration of photoreceptor cells in the retina, causing vision loss.
Genetic Transmission Usher syndrome is inherited in an autosomal recessive pattern. This means:
Both parents must carry a copy of the mutated gene to pass it on
Each child of carrier parents has a 25% chance of inheriting the condition
At least 11 genes have been associated with different types of Usher syndrome
Types: There are three main clinical types:
Type 1: Profound hearing loss or deafness at birth
Severe balance problems from birth – children often have delayed motor development and don’t walk until 18 months or later
Vision problems usually begin around age 10 or in early teens
Night blindness and loss of peripheral vision due to retinitis pigmentosa (RP)
Type 2: Moderate to severe hearing loss at birth
Normal balance
Vision problems begin in late teens or early 20s
Night blindness and gradual loss of peripheral vision due to RP, but progression is typically slower than in Type 1
Central vision often retained into adulthood
Type 3: Born with normal hearing and near-normal balance
Progressive hearing loss starting in childhood or teens
Vision loss begins in teens or early adulthood
Balance may deteriorate over time
About 50% experience balance problems
Signs and Symptoms
Hearing loss (congenital or progressive)
Progressive vision loss due to RP (night blindness, peripheral vision loss)
Balance problems (mainly in Type 1)
Delays in motor development in children (Type 1)
Potential Treatment Options While there is no cure, management focuses on:
Hearing aids or cochlear implants for hearing loss
Low vision aids and mobility training for vision loss
Vestibular rehabilitation for balance issues
Genetic counseling for families for future conception
Emerging therapies like gene therapy are under investigation
References
Arias-Peso, B., Calero-Ramos, M. L., López-Ladrón García de la Borbolla, C., López-Domínguez, M., Morillo-Sánchez, M. J., Méndez-Martínez, S., Sánchez-Gómez, S., & Rodríguez-de-la-Rúa, E. (2024). Multidisciplinary approach to inherited causes of dual sensory impairment. Graefe’s Archive of Clinical & Experimental Ophthalmology, 262(3), 701–715. https://doi.org/10.1007/s00417-023-06153-7
Mahmood, R., Mahmood, F., Faisal, M. N., Mahmood, A., Muzaffar, H., Mahmood, M., Abbas, G., Mahmood, T., & Arshad, M. (2022). Usher Syndrome and Its Genetic Characterization. Pakistan Journal of Science, 74(5), 392–402
Miyoshi, T., Belyantseva, I. A., Sajeevadathan, M., & Friedman, T. B. (2024). Pathophysiology of human hearing loss associated with variants in myosins. Frontiers in Physiology, 01-18. https://doi.org/10.3389/fphys.2024.1374901
DISCLAIMER
The Site may contain (or you may be sent through the Site) links to other websites or content belonging to or originating from third parties or links to websites and features in banners or other advertising. Such external links are not investigated, monitored, or checked for accuracy, adequacy, validity, reliability, availability, or completeness by us. WE DO NOT WARRANT, ENDORSE, GUARANTEE, OR ASSUME RESPONSIBILITY FOR THE ACCURACY OR RELIABILITY OF ANY INFORMATION OFFERED BY THIRD-PARTY WEBSITES LINKED THROUGH THE SITE OR ANY WEBSITE OR FEATURE LINKED IN ANY BANNER OR OTHER ADVERTISING. WE WILL NOT BE A PARTY TO OR IN ANY WAY BE RESPONSIBLE FOR MONITORING ANY TRANSACTION BETWEEN YOU AND THIRD-PARTY PROVIDERS OF PRODUCTS OR SERVICES.
PROFESSIONAL DISCLAIMER
The Site cannot and does not contain medical/health advice. The medical/health information is provided for general informational and educational purposes only and is not a substitute for professional advice. Accordingly, before taking any actions based upon such information, we encourage you to consult with the appropriate professionals. We do not provide any kind of medical/health advice. THE USE OR RELIANCE OF ANY INFORMATION CONTAINED ON THE SITE IS SOLELY AT YOUR OWN RISK.
AFFILIATES DISCLAIMER
The Site may contain links to affiliate websites, and we receive an affiliate commission for any purchases made by you on the affiliate website using such links.
We are a participant in the Amazon Services LLC Associates Program, an affiliate advertising program designed to provide a means for us to earn advertising fees by linking to Amazon.com and affiliated websites. This disclaimer was created using Termly’s Disclaimer Generator.
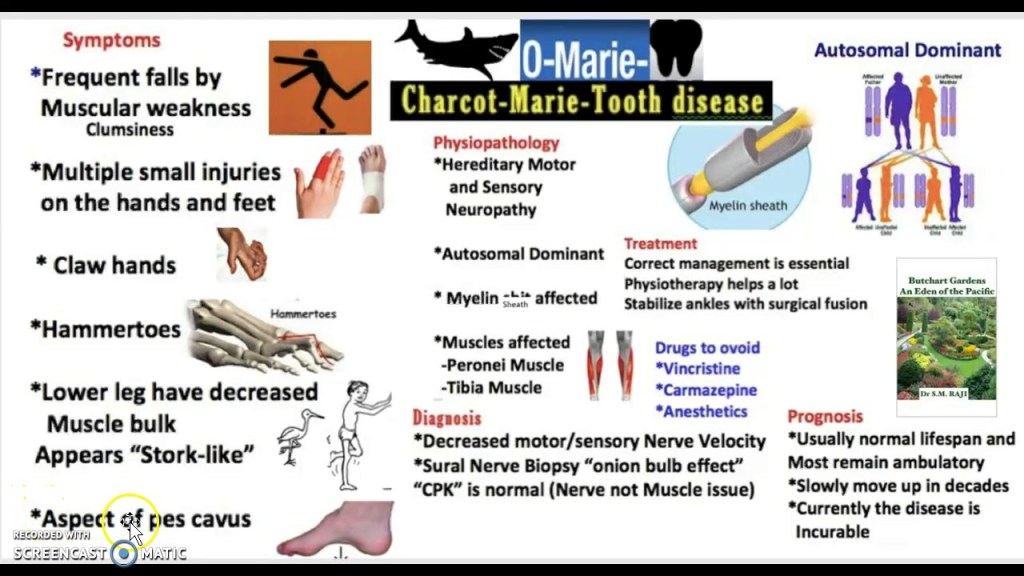
Charcot-Marie-Tooth (CMT) disease is a group of inherited peripheral neuropathies affecting motor and sensory nerves. CMT can cause a a child to lose feeling and movement in their extremities. Sometimes this disease affects speech, breathing and swallowing. With this condition, the peripheral nerves (the nerves outside the brain and spinal cord) don’t work properly.
Pathophysiology
CMT results from mutations in genes involved in the structure and function of peripheral nerves. The pathophysiology varies depending on the specific type:
Demyelinating forms (CMT1, CMT4, CMTX): Mutations affect Schwann cell function, leading to defective myelin production or maintenance. This results in reduced nerve conduction velocities.
Axonal forms (CMT2): Mutations primarily affect axonal structure or function, leading to axonal degeneration.
Intermediate forms: Show features of both demyelinating and axonal neuropathies.
The common end result is progressive axonal loss, leading to muscle weakness and sensory deficits.
Genetic Transmission
CMT can be inherited in several patterns:
Autosomal dominant: Most common in CMT1 and CMT2
Autosomal recessive: Seen in some forms of CMT4
X-linked: CMTX1 (mutations in GJB1 gene)
The most common mutations involve:
PMP22 gene duplication (CMT1A)
MPZ gene mutations (CMT1B)
GJB1 gene mutations (CMTX1)
MFN2 gene mutations (CMT2A)
X-linked CMT (CMTX) has a unique inheritance pattern compared to other forms of CMT
Inheritance through females
When a female has X-linked CMT, each of her children (regardless of gender) has a 50% chance of inheriting the CMT-causing mutation.
This is because females have two X chromosomes, and can pass either the affected or unaffected X chromosome to their children.
Inheritance through males
When a male has X-linked CMT, all of his daughters will inherit the CMT-causing mutation.
None of his sons will inherit the CMT-causing mutation.
This is because males have one X and one Y chromosome. They always pass their X chromosome (which carries the mutation) to their daughters and their Y chromosome to their sons.
Key points
X-linked CMT can be inherited through both males and females, but the pattern differs.
Females can pass it to children of either gender.
Males will only pass it to their daughters, not their sons.
The inheritance pattern is different from autosomal dominant or recessive forms of CMT.
Severity
Males with X-linked CMT typically have more severe symptoms than females.
Females may have milder symptoms or be asymptomatic carriers due to having two X chromosomes (one normal, one with the mutation).
In summary, X-linked CMT can be inherited through both males and females, but the inheritance pattern and likelihood of passing on the condition differs based on the parent’s sex.
Signs and Symptoms
Motor symptoms:
Progressive distal muscle weakness and atrophy
Foot deformities (pes cavus, hammer toes)
Difficulty walking, frequent tripping
Hand weakness and atrophy in later stages
Sensory symptoms:
Reduced sensation in feet and hands
Neuropathic pain in some cases
Other features:
Reduced or absent deep tendon reflexes
Scoliosis in some patients
Progression of CMT can be Quite Different for Different People
The progression of CMT syndrome can vary significantly between individuals, even within the same family. Here are some key points about how the progression can differ:
Age of onset: Symptoms typically appear in adolescence or early adulthood, but can start anytime from early childhood to late adulthood. Some individuals may not realize they have CMT until later in life due to very mild symptoms.
Rate of progression: CMT is generally slowly progressive, but the rate can vary. Some people experience rapid progression, while others have very slow progression over decades.
Severity of symptoms: The severity ranges from mild cases with minimal impairment to severe cases causing significant disability. Most people fall in the moderate range.
Distribution of symptoms: While CMT typically affects the feet and lower legs first, some individuals may experience hand and arm involvement earlier or more severely than others.
Types of symptoms: The balance between motor and sensory symptoms can differ. Some people may have more muscle weakness, while others experience more sensory loss.
Specific impairments: The development of foot deformities, hand dexterity issues, or breathing problems can vary widely between individuals.
Impact on daily activities: The effect on walking, balance, fine motor skills, and overall quality of life differs for each person.
Genetic factors: Different genetic mutations associated with CMT can lead to variations in disease progression and severity.
Environmental factors: Lifestyle, overall health, and environmental factors may influence how the disease progresses in different individuals.
Response to interventions: The effectiveness of treatments and interventions can vary between individuals, affecting the overall course of the disease.
It’s important to note that even within families carrying the same genetic mutation, there can be significant variability in how CMT progresses. Regular monitoring and personalized management are crucial due to this variability in progression.
Treatment Plans
There is no cure for CMT, so treatment focuses on symptom management and supportive care including physical therapy, occupational therapy, orthopedic interventions, pain management, genetic counseling for parents, and regular monitoring. Physical and occupational therapy might involve stretching and strengthening exercises, gait training, use of adaptive devices to improve hand and leg function, and strategies for regular activities of daily living (such as dressing, walking, grooming, toileting). Orthopedic interventions might include ankle-foot braces (also called ankle-foot orthoses or AFOs) for foot drop, or orthopedic surgery for deformities or scoliosis. Pain management can include over the counter pain medications, alternative pain therapy, or narcotics. Adults and parents of children with CMT can undergo genetic counseling. Finally, patients will be monitored for disease progression and deterioration.
References
Aldihan, K. A., AlRashedi, M. J., Helayel, H. B., AlMutlak, M., & Hameed, S. T. (2023). Severe Dry Eye Disease in Charcot-Marie-Tooth Disease: A Comprehensive Case Report. American Journal of Case Reports, 24, 1–4. https://doi.org/10.12659/AJCR.941094
Choi, J. E., Seol, H. Y., Seok, J. M., Hong, S. H., Choi, B. ‐O., & Moon, I. J. (2020). Psychoacoustics and neurophysiological auditory processing in patients with Charcot‐Marie‐Tooth disease types 1A and 2A. European Journal of Neurology, 27(10), 2079–2088. https://doi.org/10.1111/ene.14370
Silsby, M., Yiannikas, C., Fois, A. F., Kennerson, M. L., Kiernan, M. C., Fung, V. S. C., & Vucic, S. (2024). Upper and lower limb tremor in Charcot-Marie-Tooth neuropathy type 1A and the implications for standing balance. Journal of Neurology, 271(4), 1776–1786. https://doi.org/10.1007/s00415-023-12124-z
Waldman, L. E., Michalski, M. P., Pfeffer, G. B., Giaconi, J. C., & Learch, T. J. (2023). Charcot-Marie-Tooth Disease of the Foot and Ankle: Imaging Features and Pathophysiology. Radiographics, 43(4). https://doi.org/10.1148/rg.220114
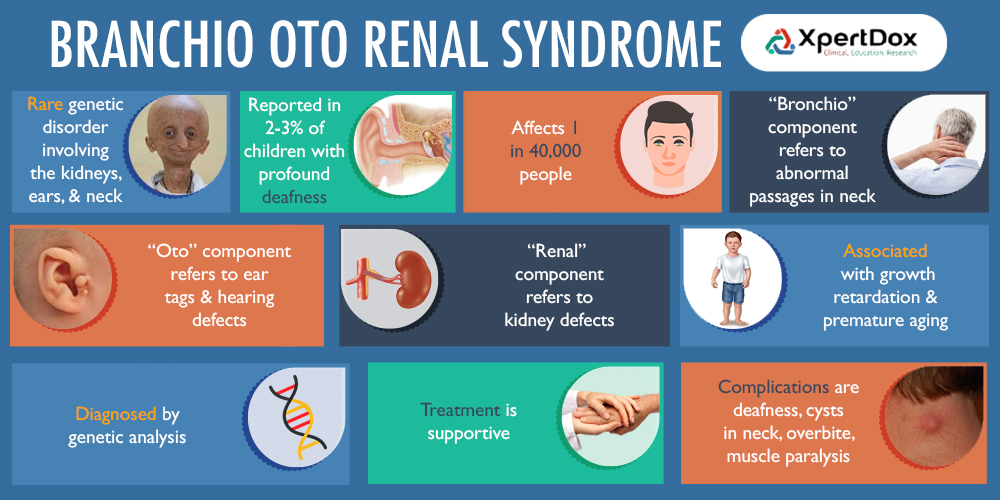
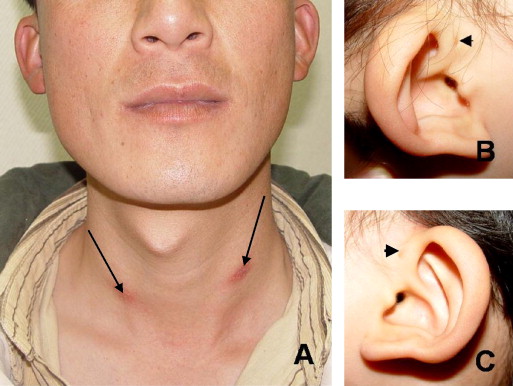
Branchio-Oto-Renal (BOR) syndrome is a genetic disorder affecting the development of the neck, ears, and kidneys.
Pathophysiology
BOR syndrome results from mutations in genes involved in embryonic development, primarily EYA1, SIX1, and SIX5. These genes play crucial roles in the development of the branchial arches, otic vesicles, and kidneys. Mutations disrupt normal tissue formation, leading to the characteristic features of BOR syndrome.
Genetic Transmission
BOR syndrome is primarily inherited in an autosomal dominant pattern. This means that only one copy of the mutated gene is needed to cause the disorder’ an affected individual has a 50% chance of passing the mutation to each offspring; and the condition can affect both males and females equally.
Some key points about genetic transmission
EYA1 mutations account for about 40% of BOR syndrome cases.
SIX1 and SIX5 mutations are less common causes.
Some cases may result from de novo mutations.
Signs and Symptoms
BOR syndrome exhibits variable expressivity, even within families. Common features include branchial anomalies that can include branchial cleft cysts or fistulae in the neck. Otic (ear) abnormalities such as hearing loss (sensorineural, conductive, or mixed), malformations of outer, middle, or inner ear, and/or preauricular pits or tags. Renal (kidney) abnormalities ranging from mild (e.g., abnormal kidney shape) to severe (e.g., renal agenesis) which can progress to end-stage renal disease later in life. Other possible features can also occur including but not limited to facial abnormalities (e.g., long, narrow face), cleft palate, or lacrimal duct stenosis.
Treatment Plans
There is no cure for BOR syndrome, so treatment focuses on managing symptoms and complications:
Hearing management:
Regular audiological assessments
Hearing aids or cochlear implants as needed
Speech and language therapy
Branchial anomalies:
Surgical removal of cysts or repair of fistulae if causing problems
Renal care:
Regular monitoring of kidney function
Management of hypertension if present
Dialysis or kidney transplantation for end-stage renal disease
Other interventions:
Surgical correction of ear malformations if desired
Treatment of associated conditions (e.g., cleft palate repair)
Genetic counseling for affected individuals and families
References
Biggs, K., Crundwell, G., Metcalfe, C., Muzaffar, J., Monksfield, P., & Bance, M. (2022). Anatomical and audiological considerations in branchiootorenal syndrome: A systematic review. Laryngoscope Investigative Otolaryngology, 7(2), 540–563. https://doi.org/10.1002/lio2.749
Cacciatori, E., Aleo, S., Scuvera, G., Rigon, C., Marchisio, P. G., Cassina, M., & Milani, D. (2022). From clinical to molecular diagnosis: relevance of diagnostic strategy in two cases of branchio-oto-renal syndrome – case report. Italian Journal of Pediatrics, 48(1), 1–6. https://doi.org/10.1186/s13052-022-01369-5
Peusner, K. D., Bell, N. M., Hirsch, J. C., Beraneck, M., & Popratiloff, A. (2021). Understanding the Pathophysiology of Congenital Vestibular Disorders: Current Challenges and Future Directions. Frontiers in Neurology, 12, 1–10. https://doi.org/10.3389/fneur.2021.708395
Yalcouyé, A., Traoré, O., Diarra, S., Schrauwen, I., Esoh, K., Kadlubowska, M. K., Bharadwaj, T., Adadey, S. M., Kéita, M., Guinto, C. O., Leal, S. M., Landouré, G., & Wonkam, A. (2022). A monoallelic variant in EYA1 is associated with Branchio‐Otic syndrome in a Malian family. Molecular Genetics & Genomic Medicine, 10(7), 1–8. https://doi.org/10.1002/mgg3.1995
Alport syndrome is a genetic disorder that affects the basement membranes of the kidneys, eyes, and inner ears. Mutations in your collagen genes cause Alport syndrome. Symptoms include blood and protein in your urine, hearing, and vision loss. It may also cause kidney failure.
Pathophysiology
Alport syndrome is caused by mutations in the COL4A3, COL4A4, or COL4A5 genes, which encode for type IV collagen. This collagen is a crucial component of basement membranes, particularly in the glomeruli of the kidneys. The mutations lead to abnormal collagen IV formation, resulting in:
Progressive damage to the glomerular basement membrane (GBM)
Impaired filtration in the kidneys
Structural changes in the cochlea and eyes
There are three main genetic types of Alport syndrome
X-linked Alport syndrome (XLAS):
Females develop kidney failure less frequently and more slowly.
This is the most common form, accounting for about 80-85% of cases.
It is caused by mutations in the COL4A5 gene located on the X chromosome.
Males are typically more severely affected than females.
Without treatment, about 90% of males develop kidney failure by age 40.
Autosomal recessive Alport syndrome (ARAS):
Both copies of the abnormal gene are needed to cause this type.
This accounts for about 10-15% of cases.
It is caused by mutations in both copies of either the COL4A3 or COL4A4 genes.
Both parents carry one abnormal gene copy and pass it to the child.
Autosomal dominant Alport syndrome (ADAS):
Only one copy of the abnormal gene is needed to cause the disease.
This is the rarest form.
It is caused by a mutation in one copy of either the COL4A3 or COL4A4 gene.
Only one parent needs to have and pass on the abnormal gene for the child to be affected.
All three types affect the COL4A3, COL4A4, or COL4A5 genes, which code for type IV collagen chains in basement membranes of the kidneys, cochlea, and eyes. The different inheritance patterns and specific genes involved determine the type of Alport syndrome. Genetic testing can help confirm the diagnosis and determine the specific type.
Signs and Symptoms
Kidney-related:
Hematuria (blood in urine) – often the earliest sign
Proteinuria (protein in urine)
Progressive decline in kidney function
Hypertension
Eventually, end-stage renal disease (ESRD)
Hearing-related:
Progressive sensorineural hearing loss, typically affecting high frequencies first
Eye-related:
Anterior lenticonus (cone-shaped protrusion of the lens)
Retinal abnormalities (flecks)
Rarely, corneal dystrophy
X-linked Alport syndrome (XLAS) and autosomal recessive Alport syndrome (ARAS) typically manifest differently:
Severity and progression in males vs females:
In XLAS, males are typically more severely affected than females. Without treatment, about 90% of males develop kidney failure by age 40. Females generally have less severe, more slowly progressing symptoms.
In ARAS, both males and females are affected with equal frequency and severity. Both typically experience kidney failure by around age 20.
Inheritance pattern:
XLAS can appear to skip generations, particularly when there’s an affected female with only hematuria. The father of an affected individual may have hematuria.
ARAS typically manifests within a single generation, affecting siblings but not parents.
Onset of symptoms:
In XLAS, males often develop renal symptoms between ages 20-30 (juvenile form), though some may develop renal insufficiency after age 30 (adult form).
ARAS tends to progress more rapidly, with kidney failure typically occurring by the end of the second decade of life.
Extra-renal manifestations:
In XLAS, females rarely exhibit hearing loss or ocular abnormalities, which are more characteristic in affected males.
In ARAS, both males and females commonly experience the full spectrum of symptoms, including renal failure, hearing loss, and ocular abnormalities.
Diagnosis in females:
XLAS can be more challenging to diagnose in females due to variable expression.
ARAS presents similarly in both males and females, potentially making diagnosis more straightforward.
While both types share common features like hematuria, proteinuria, and progression to kidney failure, the key differences lie in the severity, age of onset, and gender-specific manifestations, particularly in XLAS.
Treatment Plans
Kidney management:
ACE inhibitors or angiotensin receptor blockers (ARBs) to reduce proteinuria and slow kidney disease progression
Blood pressure control
Dietary modifications (low sodium, controlled protein intake)
Regular monitoring of kidney function
Dialysis or kidney transplantation for ESRD
Hearing management:
Regular audiological assessments
Hearing aids when necessary
Eye care:
Regular ophthalmological examinations
Treatment of specific eye complications as needed
Genetic counseling for affected individuals and families
Supportive care and management of complications
References
Eriksen, K. O., & Jørstad, Ø. K. (2020). Multiple Vitelliform Lesions as a Retinal Manifestation of Alport Syndrome. Case Reports in Ophthalmology, 11(1), 79–84. https://doi.org/10.1159/000505948
Zhang, Hongwen, et al. “Combined Alport Syndrome, Klinefelter Syndrome and Fanconi Syndrome in a Chinese Boy.” Nephrology, vol. 28, no. 5, May 2023, pp. 272–75. EBSCOhost, https://doi.org/10.1111/nep.14152.
Raquel Martínez-Pulleiro, María García-Murias, Manuel Fidalgo-Díaz, & Miguel Ángel García-González. (2021). Molecular Basis, Diagnostic Challenges and Therapeutic Approaches of Alport Syndrome: A Primer for Clinicians. International Journal of Molecular Sciences, 22(20), 11063. https://doi.org/10.3390/ijms222011063
Misato Kamura, Tomohiko Yamamura, Kohei Omachi, Mary Ann Suico, Kandai Nozu, Shota Kaseda, Jun Kuwazuru, Tsuyoshi Shuto, Kazumoto Iijima, & Hirofumi Kai. (2020). Trimerization and Genotype–Phenotype Correlation of COL4A5 Mutants in Alport Syndrome. Kidney International Reports, 5(5), 718–726. https://doi.org/10.1016/j.ekir.2020.01.008
Waardenburg syndrome is a rare genetic condition caused by a group of genetic conditions that cause congenital hearing loss, pigmentation deficiencies, and specific facial features.
Common Features
Includes bright blue eyes or one blue and one brown eye, a white forelock, or patches of light skin.
Causes
Mutations in genes affecting neural crest cells during embryonic development, with PAX3, MITF, and SOX10 being notable ones for different types.
Pathophysiology
Waardenburg syndrome (WS) is a genetic disorder caused by mutations in genes involved in the development and migration of neural crest cells during embryogenesis. The main genes implicated are PAX3, MITF, SOX10, EDNRB, and EDN3. These mutations lead to abnormal development of melanocytes, resulting in pigmentation abnormalities and hearing loss. The specific pathophysiology varies by WS type:
WS1 and WS3: Primarily caused by PAX3 mutations, affecting melanocyte development and migration
WS2: Mostly due to MITF mutations, impacting melanocyte survival and function
WS4: Involves SOX10, EDNRB, or EDN3 mutations, affecting both melanocytes and enteric neurons
The abnormal melanocyte development and distribution leads to the characteristic pigmentary changes and sensorineural hearing loss seen in WS.
Congenital Transmission:
Waardenburg syndrome is primarily inherited in an autosomal dominant pattern, meaning only one copy of the altered gene is needed to cause the disorder. However, the specific genetic transmission can vary depending on the type of Waardenburg syndrome:
Waardenburg syndrome types 1 and 3:
Caused by mutations in the PAX3 gene
Inherited in an autosomal dominant pattern
A person with the mutation has a 50% chance of passing it on to each child
Waardenburg syndrome type 2:
Most commonly caused by mutations in the MITF gene, but can also be caused by mutations in other genes like SOX10
Inherited in an autosomal dominant pattern
Some cases may occur sporadically due to new mutations
Waardenburg syndrome type 4:
Can be caused by mutations in several genes including SOX10, EDNRB, and EDN3
Inheritance pattern can vary:
SOX10 mutations are typically autosomal dominant
EDNRB and EDN3 mutations are usually autosomal recessive, requiring two copies of the altered gene
Key points about the congenital transmission of Waardenburg syndrome:
Variable expressivity: The severity and specific features can vary widely, even within the same family.
Incomplete penetrance: Some individuals who inherit the mutation may not show symptoms.
Sporadic cases: In some instances, the syndrome can occur due to new mutations in individuals with no family history of the disorder.
Genetic testing: Can be used to confirm the diagnosis and identify the specific gene mutation, which is helpful for genetic counseling.
Prenatal testing: Is possible for some types of Waardenburg syndrome if the specific mutation in the family is known.
Autosomal dominant inheritance is a pattern of genetic transmission where:
Only one copy of the mutated gene is needed to cause the disorder or trait.
An affected individual has a 50% chance of passing the mutated gene to each offspring, regardless of the child’s sex.
Both males and females are equally likely to be affected and to transmit the trait.
Expressivity can be variable, with affected individuals showing different degrees of severity.
Signs and Symptoms:
The main clinical features of WS include:
Pigmentary abnormalities:
White forelock (poliosis)
Premature graying of hair
Heterochromia iridis (different colored irises)
Patchy depigmentation of skin (leukoderma)
Hearing loss: Congenital sensorineural hearing loss, usually bilateral
Craniofacial features:
Dystopia canthorum (wide-set inner corners of eyes) in WS1 and WS3
Broad nasal root
Synophrys (connected eyebrows)
Additional features in specific types:
WS4: Hirschsprung disease is a condition that affects the large intestine (colon) and causes problems with passing stool. The condition is present at birth (congenital) as a result of missing nerve cells in the muscles of the baby’s colon. Without these nerve cells stimulating gut muscles to help move contents through the colon, the contents can back up and cause blockages in the bowel.
WS3: Upper limb abnormalities. The upper limbs may appear underdeveloped with flexion contractures, fusion of the carpal bones and sometimes syndactyly
The presentation can be highly variable, even within families.
Treatment Plans:
There is no cure for WS, so treatment focuses on managing symptoms and associated complications:
Hearing management:
Early hearing screening and intervention
Hearing aids or cochlear implants for hearing loss
Speech and language therapy
Ophthalmological care:
Regular eye exams
Correction of refractive errors
Monitoring for glaucoma
Dermatological management:
Sun protection for depigmented areas
Cosmetic treatments (e.g., hair dyes, skin camouflage) if desired
Genetic counseling for affected individuals and families
Specific management for associated conditions:
Surgical intervention for Hirschsprung disease in WS4
Orthopedic management for limb abnormalities in WS3
Supportive care and developmental assessments, especially for children with hearing loss
References:
Chen, L., Wang, L., Chen, L., Wang, F., Ji, F., Sun, W., Zhao, H., Han, W., & Yang, S. (2020). Transcript Profiles of Stria Vascularis in Models of Waardenburg Syndrome. Neural Plasticity, 1–9. https://doi.org/10.1155/2020/2908182
Lee, C., Lo, M., Chen, Y., Lin, P., Hsu, C., Chen, P., Wu, C., & Hsu, J. S. (2022). Identification of nine novel variants across PAX3, SOX10, EDNRB, and MITF genes in Waardenburg syndrome with next‐generation sequencing. Molecular Genetics & Genomic Medicine, 10(12), 1–13. https://doi.org/10.1002/mgg3.2082.
Sun, F., Xiao, M., Ji, D., Zheng, F., & Shi, T. (2024). Deciphering potential causative factors for undiagnosed Waardenburg syndrome through multi-data integration. Orphanet Journal of Rare Diseases, 19(1), 226. https://doi.org/10.1186/s13023-024-03220-y
Li, Y., Chen, Y., Sun, Y., Li, S., Dong, L., Li, Z., & Shen, G. (2024). Waardenburg syndrome type 2 with a de novo variant of the SOX10 gene: a case report. BMC Medical Genomics, 17(1), 104. https://doi.org/10.1186/s12920-024-01877-9
Lin, P.-A., Hung, J.-H., & Huang, Y.-H. (2024). Heterochromia caused by Waardenburg syndrome in a 2-month-old infant. CMAJ : Canadian Medical Association Journal = Journal de l’Association Medicale Canadienne, 196(9), E296. https://doi.org/10.1503/cmaj.231616
Age-related hearing loss is common in those persons over 65 years of age. Nearly 33% of persons over 65 years of age experience this type of hearing loss. Age-related hearing loss is typically a high-frequency loss that occurs progressively but slowly throughout adulthood. While age-related hearing loss begins in the high frequencies it slowly progresses to lower frequencies with time. This study examined the relationship between age-related hearing loss and anxiety in those over 65 years of age.
Sixty-seven persons with age-related hearing loss and 68 normal-hearing controls participated in this cross-sectional study. The criteria to be included in the age-related hearing loss group was a four-frequency pure tone average of >25 decibels hearing level in the better hearing ear. All participants had three-dimensional magnetic resonance imaging (MRI), pure tone audiometric testing, and anxiety and depression scales.
The findings included a decrease in grey matter volume in 20 brain regions in the age-related hearing loss group. In addition, a positive correlation was found between high-frequency pure tone average and anxiety scores in the age-related hearing loss group. No relationships were found between depression and either gray matter volume or high-frequency hearing loss.
Ma, W., Zhang, Y., Li, X., Liu, S., Gao, Y., Yang, J., Xu, L., Liang, H., Ren, F., Gao, F., & Wang, Y. (2022). High-Frequency Hearing Loss Is Associated with Anxiety and Brain Structural Plasticity in Older Adults. Frontiers in aging neuroscience, 14, 821537. https://doi.org/10.3389/fnagi.2022.821537






























Leave a comment