
Pulmonary fibrosis (PF) is a progressive lung disease characterized by scarring (fibrosis) of the lung tissue, leading to impaired lung function and breathing difficulties. This article provides an overview of PF, including its prevalence, epidemiology, pathophysiology, clinical presentation, and current treatment approaches.
Prevalence and Incidence in the US
Accurate estimates of PF prevalence and incidence are challenging due to varying diagnostic criteria and underdiagnosis. However, recent studies suggest:
- Prevalence: Approximately 200,000 people in the US are living with PF, with idiopathic pulmonary fibrosis (IPF) being the most common form.
- Incidence: The annual incidence of IPF is estimated at 6.8-16.3 cases per 100,000 person-years.
Epidemiology
PF affects people of all ages, but risk increases with age. Key epidemiological factors include:
Most commonly diagnosed in individuals over 50, with peak incidence between 60-70 years. PF is slightly more common in males than females. Some studies suggest higher prevalence in non-Hispanic whites. Smoking, environmental/occupational exposures, genetic predisposition, and certain medical conditions such a Gastroesophageal reflux disease (GERD) are the most common risk factors for PF. Using narrow case definitions, prevalence ranges from 14 to 27.9 cases per 100,000 population. Overall prevalence is estimated to be around 13 to 20 cases per 100,000 people. About 100,000 people are affected by idiopathic pulmonary fibrosis (IPF) in the United States. Approximately 30,000 to 40,000 new cases of IPF are diagnosed each year. Some studies suggest the incidence and prevalence of IPF may be increasing in recent years.
It’s important to note that epidemiological data on pulmonary fibrosis can be challenging to obtain due to changes in diagnostic criteria over time, potential underdiagnosis, and variations in study methodologies. The figures provided represent the best available estimates based on current research, but there is still uncertainty around the true epidemiology of this condition in the US.
Pathophysiology
The pathophysiology of PF involves complex interactions between genetic susceptibility, environmental factors, and aberrant wound healing responses. Key mechanisms include:
- Repetitive alveolar epithelial cell injury
- Abnormal activation of fibroblasts and myofibroblasts
- Excessive deposition of extracellular matrix proteins
- Dysregulated immune responses
- Oxidative stress and mitochondrial dysfunction
These processes lead to progressive scarring of lung tissue, reducing lung compliance and impairing gas exchange.


Signs and Symptoms
Common signs and symptoms of PF include:
- Dyspnea (shortness of breath), initially with exertion and progressing to occur at rest
- Dry, persistent cough
- Fatigue and weakness
- Unintended weight loss
- Clubbing of fingers and toes
- Fine crackles on lung auscultation
- Chest discomfort
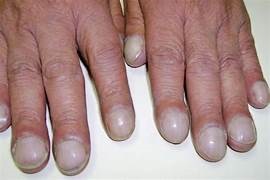
This image shows advanced finger clubbing. Clubbing occurs in individuals who have chronic, long-term periods of hypoxia. The exact mechanism for this is unknown.
As the disease progresses, patients may develop complications such as pulmonary hypertension, right heart failure, and acute exacerbations.
Treatment
While there is no cure for PF, several treatment options aim to slow disease progression, manage symptoms, and improve quality of life:
- Antifibrotic medications: Pirfenidone and nintedanib have shown efficacy in slowing lung function decline in IPF.
- Anti-inflammatory agents: Corticosteroids and immunosuppressants may be used in some forms of PF.
- Oxygen therapy: Supplemental oxygen can improve exercise tolerance and quality of life.
- Pulmonary rehabilitation: Exercise training and education programs can enhance functional capacity and well-being.
- Lung transplantation: Considered for select patients with advanced disease.
- Symptom management: Treatments for cough, breathlessness, and anxiety/depression.
- Supportive care: Including vaccinations, management of comorbidities, and palliative care when appropriate.
Recent research is exploring novel therapeutic targets and combination therapies to further improve outcomes in PF.
Conclusion
Pulmonary fibrosis remains a challenging condition with significant morbidity and mortality. Ongoing research into its pathophysiology and novel treatment approaches offers hope for improved management and outcomes for patients with this devastating disease.
References
- Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941-1952. doi:10.1016/S0140-6736(17)30866-8
- Martinez FJ, Collard HR, Pardo A, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017;3:17074. doi:10.1038/nrdp.2017.74
- Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med. 2018;378(19):1811-1823. doi:10.1056/NEJMra1705751
- Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198(5):e44-e68. doi:10.1164/rccm.201807-1255ST
- Selman M, Pardo A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell Signal. 2020;66:109482. doi:10.1016/j.cellsig.2019.109482
- Kolb M, Vasakova M. The natural history of progressive fibrosing interstitial lung diseases. Respir Res. 2019;20(1):57. doi:10.1186/s12931-019-1022-1
- Maher TM, Strek ME. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respir Res. 2019;20(1):205. doi:10.1186/s12931-019-1161-4
