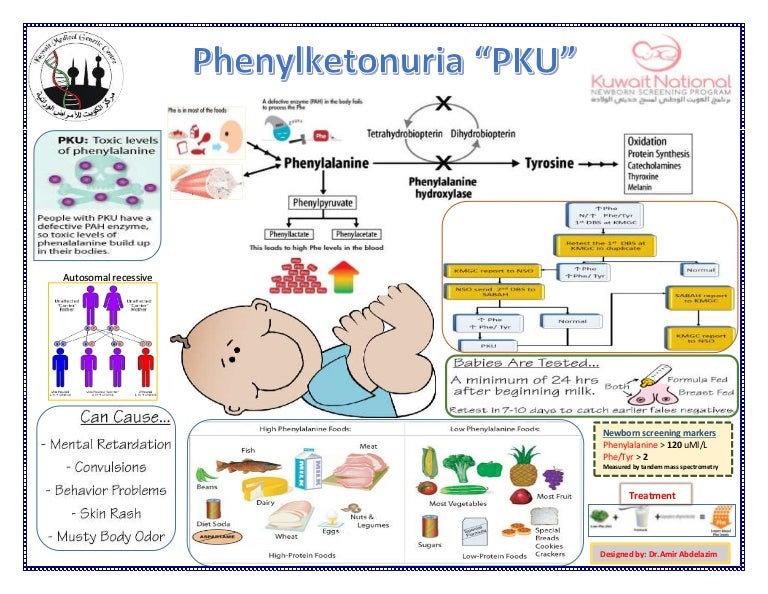
Phenylketonuria (PKU) is a rare, inherited metabolic disorder that necessitates vigilant nursing assessment to manage effectively and prevent complications. Nursing assessment for PKU involves a thorough examination of the patient’s medical history, physical condition, and dietary habits. The cornerstone of the assessment is early detection, usually through newborn screening, as prompt intervention is vital to prevent intellectual disabilities and other associated complications.
Physical Characteristics
One distinctive feature of the PKU phenotype is intellectual disability. Elevated levels of phenylalanine in the blood, resulting from the inability to break down this amino acid, can lead to damage to the developing brain, particularly affecting cognitive function. If left untreated, individuals with PKU may experience learning difficulties, developmental delays, and intellectual impairment.
Another notable aspect of the PKU phenotype is the potential for neurological symptoms. These may include seizures, tremors, or other movement disorders. The impact on the nervous system is a consequence of the toxic effects of high phenylalanine levels on the developing brain and underscores the importance of early detection and intervention.
Physical characteristics associated with PKU may include fair skin, light hair, and blue eyes. These features result from the influence of phenylalanine on the production of melanin, the pigment responsible for skin, hair, and eye color. The phenylalanine-restricted diet that individuals with PKU must adhere to can influence these physical traits, as it limits the intake of phenylalanine-containing foods.
In addition to cognitive and neurological effects, individuals with PKU may exhibit behavioral and psychiatric symptoms. These can include mood disorders, attention deficits, and behavioral challenges. The interplay of elevated phenylalanine levels and their impact on neurotransmitter function in the brain contributes to these behavioral aspects of the PKU phenotype.
It’s important to note that the severity of the PKU phenotype can vary depending on the level of phenylalanine restriction achieved through dietary management and other therapeutic interventions. Early diagnosis through newborn screening and prompt initiation of a phenylalanine-restricted diet are crucial for minimizing the impact of PKU on the individual’s phenotype.

Genetic Transmission
A critical component of the nursing assessment for PKU is understanding the patient’s genetic background and family history. Since PKU is an autosomal recessive disorder, obtaining information about the presence of the condition in the family is crucial for assessing the risk of inheritance in subsequent generations. Knowledge of the family’s ability to adhere to treatment and dietary restrictions is also essential, as successful management of PKU relies significantly on lifestyle modifications.
The normal function of phenylalanine hydroxylase is to convert the amino acid phenylalanine to another amino acid, tyrosine, in the body. However, individuals with PKU have mutations in both copies of the PAH gene, leading to a deficiency or complete absence of functional phenylalanine hydroxylase. As a result, phenylalanine accumulates in the blood and body tissues, leading to the characteristic symptoms and complications associated with PKU.
The genetic transmission of PKU follows a predictable pattern. If both parents are carriers of a single mutated PAH gene but do not have PKU themselves (referred to as heterozygous carriers), there is a 25% chance with each pregnancy that their child will inherit two copies of the mutated gene and thus develop PKU. The probabilities for the offspring are as follows:
- 25% chance of having PKU: The child inherits one mutated gene from each parent.
- 50% chance of being a carrier: The child inherits one mutated gene from one parent but a normal gene from the other.
- 25% chance of having a normal genotype: The child inherits a normal gene from both parents and does not have PKU nor is a carrier.
It is important to note that individuals who inherit only one copy of the mutated gene (carriers) typically do not show symptoms of PKU. However, if two carriers have a child together, there is a risk that the child may inherit two mutated copies of the gene and develop PKU.
Newborn screening is a crucial component in the early detection of PKU, allowing for prompt intervention and management. By identifying infants with elevated phenylalanine levels shortly after birth, healthcare providers can implement dietary restrictions, such as a low-phenylalanine diet, to prevent the intellectual and developmental disabilities associated with untreated PKU.
The genetic transmission of PKU follows an autosomal recessive pattern, with both parents needing to be carriers for there to be a risk of their child developing the disorder. Understanding the genetic basis of PKU is essential for genetic counseling, family planning decisions, and early intervention strategies.
Management
The management of PKU involves a lifelong commitment to a carefully controlled diet low in phenylalanine. Additionally, medical formulas and supplements may be prescribed to ensure that individuals with PKU receive the necessary nutrients while adhering to dietary restrictions. Regular monitoring of blood phenylalanine levels is essential to adjust dietary interventions and prevent the harmful effects associated with elevated phenylalanine.
Physical examination plays a pivotal role in assessing the impact of PKU on the patient’s overall health. Monitoring growth and development, particularly neurodevelopmental milestones, is crucial to identify any signs of intellectual disabilities. Additionally, assessing for signs of phenylalanine toxicity, such as musty odor in breath and urine, is imperative. Neurological assessments, including coordination and muscle tone evaluations, aid in identifying any neurological deficits associated with elevated phenylalanine levels.
Regular monitoring of blood phenylalanine levels is a fundamental aspect of nursing assessment for PKU. These levels serve as a direct indicator of dietary compliance and the effectiveness of treatment. Nurses play a key role in educating patients and their families about the importance of adhering to a phenylalanine-restricted diet and regularly obtaining blood tests to maintain phenylalanine levels within the recommended range.
Nutritional assessment is a central component of PKU management. Nurses work closely with dietitians to ensure that patients receive adequate nutrition while adhering to the phenylalanine-restricted diet. This involves monitoring protein intake from natural foods and specialized medical formulas designed for individuals with PKU. The nurse’s role extends to providing ongoing education and support to patients and families in navigating the challenges of dietary restrictions and incorporating appropriate substitutes for protein-containing foods.
Psychosocial assessment is crucial in understanding the impact of PKU on the patient’s and family’s quality of life. Living with a chronic condition that requires strict dietary adherence can be challenging, and nurses play a vital role in assessing coping mechanisms, offering emotional support, and facilitating access to support groups or counseling services.
Conclusion
Nursing assessment of PKU is a comprehensive process that encompasses genetic, physical, nutritional, and psychosocial aspects. Early detection, vigilant monitoring, and effective communication with patients and their families are paramount in ensuring optimal outcomes for individuals with PKU. By addressing the multifaceted dimensions of PKU, nurses contribute significantly to the overall well-being and quality of life of those affected by this metabolic disorder.
